Bacterial DNA Persists for Extended Periods after Cell Death
Geoffrey Young, BDS,* Sally Turner, BSc, PhD,† John K. Davies, BSc, PhD,†
Göran Sundqvist, DDS, PhD,‡ and David Figdor, MDSc, FRACDS, DipEndo, PhD, FASM*†‡
Abstract
The fate of DNA from bacteria that infect the root canal but cannot survive is currently unknown, yet such information is essential in establishing the validity of polymerase chain reaction (PCR)– based identification methods for root canal samples. This in vitro study tested the hypothesis that PCR-detectable DNA from dead bacteria might persist after cell death and investigated the efficiency of sodium hypochlorite (NaOCl) as a field decontamination agent. Using heat-killed Enterococcus faecalis, the persistence of DNA encoding the 16S rRNA gene was monitored by PCR. While most probable number analysis showed an approximate 1000-fold decay in amplifiable template, E. faecalis DNA was still PCR-detectable 1 year after cell death. NaOCl (1%) eliminated amplifiable DNA within 60 seconds of exposure. Our findings also disclosed a previously overlooked problem of concentration-dependent inhibition of the PCR reaction by thiosulfate-inactivated NaOCl. These results highlight the challenges of reliably identifying the authentic living root canal flora with PCR techniques. (J Endod 2007;33:1417–1420)
Key Words
Dead bacteria, DNA, PCR, persistence, sodium hypochlorite
Every species in the oral cavity has an opportunity to enter the root canal, and while a diversity of organisms might invade, only a restricted group have the capacity to establish a viable infection (1). This is because the root canal is a unique environment that selects for a limited assortment of the oral flora (2). Advanced culture methods have been essential for characterizing the polymicrobial infection in the infected root canal (3).
A more diverse endodontic microflora than that demonstrated by culture has been described with molecular methods. With polymerase chain reaction (PCR) techniques, several culture-difficult species have been reported to be more prevalent in root canal samples than previously thought. These include spirochetes (4) and the species Tannerella forsythensis (5). In addition, analysis of endodontic samples using PCR amplification of 16S rDNA has revealed newly detected species assumed to be as-yet-uncultivable microorganisms (6).
Although molecular analysis has facilitated identification of culture-difficult species, PCR-based techniques cannot distinguish between DNA from viable or dead cells, and it is unclear whether the detected organisms truly represent the authentic living endodontic flora or rather a historical record of organisms that have entered but not survived in the root canal.
Nonsurviving species presumably disintegrate, yet remarkably little is known about the fate of their DNA in this nonvascular compartment. DNA fragments might linger and conceivably be detected and amplified by PCR. How long amplifiable DNA might persist is of considerable relevance for the validity of PCR-based methods. Under favorable conditions, DNA can be preserved for extended periods, as demonstrated by its recovery from ancient remains (7). Conversely, rapid DNA lysis has been associated with tissue storage at elevated temperature (8) and in damp (9) and biologically active environments (10). Thus, environmental conditions are likely to be critical for the rate of microbial DNA decay after cell death.
Identification of the authentic root canal flora depends on excluding non–root canal microbes and DNA; thus, thorough decontamination of the tooth and sampling field is essential before sampling the root canal. Sodium hypochlorite (NaOCl) has been advocated as a field decontamination agent (11) followed by inactivation of NaOCl with sodium thiosulfate. The thiosulfate-NaOCl reaction produces sodium salts (12) that could inhibit the PCR reaction, raising the question of possible false-negative PCR results.
There is a gap in the literature concerning the fate of DNA from microorganisms that enter the root canal and do not survive, yet cannot be eliminated by the host’s cellular defenses. The aims of this study were to test, during a 1-year experimental period, the hypothesis that PCR-detectable DNA might persist after cell death and to test current decontamination protocols for reliable recovery of PCR-based isolates.
Materials and Methods
Bacterial Strain and Culture Conditions
The bacterial strain used was Enterococcus faecalis strain JH2-2, derived from the parental strain JH2 (13). Cultures were grown overnight in 10 mL Brain Heart Infusion broth (BHI; Oxoid Ltd, Basingstoke, UK) at 37°C with shaking in air. A 2% inoculum in 50 mL BHI was then grown to the mid-exponential phase (OD600 of 0.6). Cells were harvested by centrifugation (3000g, 10 minutes), washed in sterile phosphate-buffered saline (PBS; pH 8.7), and resuspended in 25 mL PBS to a concentration of approximately 109 colony-forming units (CFU)/mL. Experiments were performed in triplicate.
Inactivation Treatment
Cell death was induced by incubation at 65°C for 2 hours. After heat treatment, cell suspensions were transferred to aerobic incubation at 37°C. To confirm cell death, a 100-µL aliquot was removed immediately after heat treatment, diluted into 10 mL BHI broth, and incubated at 37°C for 3 days before examination for turbidity. Aliquots were aseptically removed for plating and DNA extraction before heat treatment and thereafter at monthly intervals for 1 year. At each sampling time, a 1-mL aliquot was used for DNA extraction, and a 100-µL aliquot was plated onto BHI agar and incubated at 37°C for 7 days as a contamination/nonviability control.
DNA Extraction Method
Cells were harvested from 1-mL aliquots by centrifugation (12,000g, 2 minutes) and resuspended in 200 µL Gram Positive Lysis Solution with the addition of lysozyme (45 mg/mL), mutanolysin (500 U/mL), and achromopeptidase (1500 U/mL) (all from Sigma-Aldrich, St Louis, MO). After incubation at 37°C for 1.5 hours, DNA was isolated using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) following the manufacturer’s instructions. DNA was eluted into 200 µL of sterile distilled water (dH2O; pH 8.5).
PCR
The primers used are highly specific to the E. faecalis 16S rRNA gene, producing a 138 base pair (bp) amplicon (14). The primer sequences were 5?-CCGAGTGCTTGCACTCAATTGG-3’ and 5’-CTCTTAT-GCCATGCGGCATAAAC-3’. PCR was performed in a 25 µL reaction mixture containing 10 µL of extracted DNA, 0.4 µmol/L of each primer, 0.2 mmol/L of each deoxyribonucleoside triphosphate (Promega, Madison, WI), 1 UTaqDNApolymerase (Roche, Mannheim, Germany),2.5 µL of 10x PCR buffer (Roche), and dH2O. PCR amplification was performed with an Applied Biosystems 2720 (Foster City, CA) thermal cycler. Cycle parameters included an initial denaturation step at 94°C for 5 minutes, followed by 35 cycles at 94°C for 30 seconds, 60°C for 45 seconds, 72°C for 30 seconds, and a final extension step at 72°C for 7 minutes. All PCR reactions had controls. For positive controls, extracted E. faecalis DNA was used as template. For negative controls, 1 mL of PBS was subject to DNA extraction, and 10 µL of eluate was used as template.
Electrophoresis and Imaging
PCR products were analyzed by electrophoresis on 1% agarose gel stained with ethidium bromide and viewed under ultraviolet light with a Fujifilm LAS-3000 imager (Fuji, Tokyo, Japan). Reactions were positive in the presence of a 138 bp band. The size marker was lambda DNA Hind III digest (Sigma-Aldrich).
Most Probable Number Analysis
The most probable number (MPN) method of deriving population estimates in a dilution series was adopted as a semiquantitative means of documenting DNA decay (15). Ten-fold serial dilutions of each DNA extract were made with dH20, and 10 µL of each dilution was used as PCR template. After 3 replicates, the number of positive PCR reactions was scored, and a computer program (16) was used to determine the MPN and 95% confidence interval for the number of E. faecalis 16S rRNA genetic units.
Sensitivity Assays
Sensitivity of the PCR system was determined by (a) diluting a suspension of 109 CFU/mL in 10-fold series before DNA extraction and by (b) 10-fold dilution series of extracted DNA from 109 cells, with the amount of DNA estimated by measurement of absorbance at 260 nm with a NanoDrop ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, DE). The series were run in triplicate.
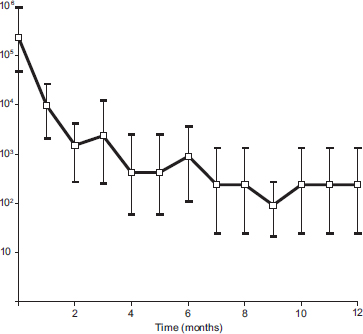
Figure 1. MPN estimate of E. faecalis 16S rRNA genetic units over time. Values are given as MPN for 3 replicates. Error bars represent the upper and lower 95% confidence limits. Value at time zero is MPN for DNA extracted from live cells.
Effect of Sodium Hypochlorite
NaOCl solutions were prepared by diluting a 1% NaOCl solution (10,000 ppm available chlorine, pH 13; Endosure, DentaLife, Croydon, Victoria, Australia) to 0.1% or 0.01%. DNA was extracted from live E. faecalis cells (approximately 109 CFU), and 10-µL aliquots of DNA suspension (approximately 100 ng/µL) were used in experiments. Ten microliters of NaOCl (1.0%, 0.1%, 0.01%) was added to 10-µL DNA suspensions and mixed by pipetting. After 60-second incubation at room temperature, NaOCl was inactivated by 10 µL sodium thiosulfate (5.0%, 0.5%, 0.05% w/v). One microliter of solution was then used as PCR template. Experiments were repeated with DNA extracted from heat-killed cells 1 month after inactivation. An additional positive control was included in NaOCl experiments to exclude PCR inhibition by thiosulfate-inactivated NaOCl. Here, NaOCl was inactivated by sodium thiosulfate before addition to DNA suspensions.
Results
Aliquots removed from cell suspensions immediately after heat treatment and incubated in BHI broth showed no turbidity, confirming an absence of viable cells. Aliquots removed for plating after treatment showed no growth, confirming maintenance of sterility. In all PCR reactions positive controls showed a 138 bp band, whereas negative controls gave no PCR signal. The DNA extraction protocol resulted in a mean detection limit of 1.1 × 103 ± 1.35 × 102 cells/mL. The mean detection limit of DNA in serial dilutions was 0.98 ± 0.03 pg.
Persistence of DNA after Cell Death
E. faecalis DNA was detected by PCR in live culture cells and in heat-killed cells. DNA from the 16S rRNA gene was detected after heat treatment and subsequent incubation for 1 year. MPN analysis revealed an approximate 1000-fold decay in the amount of amplifiable DNA during the study period (Fig. 1).
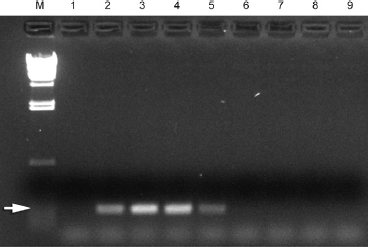
Figure 2. Inhibition of PCR by thiosulfate (5%) inactivated NaOCl (1%). Lanes M, molecular weight marker; 1, negative control; 2, E. faecalis DNA (positive control). Lanes 3–9, 0.3, 0.6, 0.9, 1.2, 1.5, 1.8, and 2.1 µL 1:1 thiosulfate/NaOCl mix, respectively. Arrow shows reaction product of 138 bp.
Effect of Sodium Hypochlorite
Experiments showed concentration-dependent inhibition of the PCR reaction by sodium thiosulfate–inactivated NaOCl. PCR gave no signal when ≥1.2 µL of thiosulfate-inactivated NaOCl (0.6 µL 1% NaOCl, 0.6 µL 5% sodium thiosulfate) was included in a 25-µL reaction mixture, whereas a positive signal was observed with ≤0.9 µL of thiosulfate-inactivated NaOCl (Fig. 2).
Treatment of DNA (live cell extract) with 1% NaOCl for 60 seconds eliminated PCR amplification of the 138-bp segment (Fig. 3). Treatment of DNA (killed cell extract) with 1% or 0.1% NaOCl for 60 seconds eliminated PCR amplification. All positive controls gave a PCR signal, confirming that the thiosulfate-inactivated NaOCl did not inhibit the reaction.
Discussion
The PCR method has been applied in detection of microbial DNA in the infected root canal, with studies confirming the presence of DNA from previously known endodontic pathogens as well as various culture-difficult species (6). The fate of DNA from bacteria that enter the root canal but cannot survive has not been previously investigated, yet such information is essential in establishing the validity of PCR-based identification methods. In this in vitro study, DNA was detectable by PCR 1 year after cell death.
Characterization of DNA decay by MPN analysis showed an approximate 1000-fold reduction in amplifiable 16S rRNA genetic units during a period of 1 year. There was a logarithmic decline in amplifiable DNA, with an early rapid decrease that subsequently appeared to plateau but remained above detection limits. These findings show that nucleic acid sequences are detectable by PCR long after cessation of bacterial cell viability.
To our knowledge, this is the first long-term study on the fate of bacterial DNA after cell death. Only a few studies with short observation times have quantified the persistence of bacterial DNA. A positive PCR signal was obtained 30 hours–18 days after cell death for Escherichia coli (17–19) and 26 days for Listeria monocytogenes (18). Persistence of amplifiable DNA varies greatly depending on the manner of cell death and the environmental milieu. Experiments assessing the impact of different stress treatments have shown that PCR gave a positive signal to cells killed by boiling, autoclaving, desiccation, starvation, ultraviolet irradiation, or ethanol treatment, yet treatment with acid or hydrogen peroxide caused rapid loss of a PCR signal (17, 18, 20). That no PCR signal was obtained from cells killed by acid or hydrogen peroxide is presumably because a low pH causes depurination of DNA, whereas hydrogen peroxide damages DNA through oxidative reactions (21). Thus, detection by PCR is possible as long as nucleic acid sequences remain intact.
The upper limit of DNA longevity after cell death is determined by spontaneous decomposition via hydrolysis, oxidation, and alkylation, as well as damage caused by ultraviolet light (21). These processes occur slowly, so that hydrated DNA is spontaneously degraded to short fragments during a period of several thousand years, although the rate of decay is profoundly affected by environmental conditions (21). Temperature, moisture, pH, oxidative agents, and mechanical stress are all important factors influencing DNA survival (22).
Deoxyribonucleases of microbial cells contribute to degradation of DNA in non-sterile environments (17). It is also known that DNA is stabilized by adsorption to minerals such as hydroxyapatite (23) where it might be protected from degradation by nucleases (24, 25). This stabilizing effect was demonstrated in a study in which mineral-bound DNA from dead bacteria was detected by PCR in a non-sterile environment after 13 weeks, whereas unbound DNA was rapidly degraded (26). Thus, in the infected root canal persistence of DNA might be influenced by various environmental factors such as destruction by nucleases and protection by mineral adsorption.
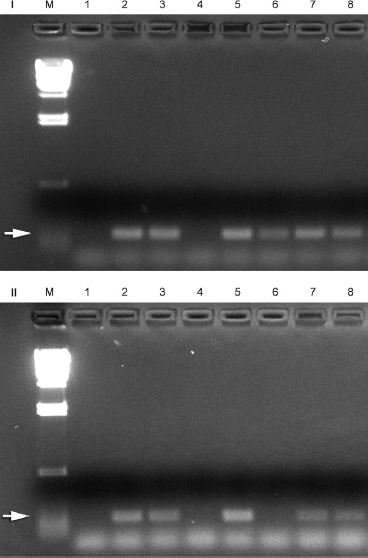
Figure 3. Effect of NaOCl on DNA detectable by PCR, extracted from live cells (I) and heat-killed cells (II). Lanes M, molecular weight marker; 1, negative control; 2, 0.33 µL E. faecalis DNA (positive control); 3, positive control (0.33 µL DNA, 0.66 µL 1:1 NaOCl [1%]/thiosulfate [5%]); 4, 60-second exposure 1% NaOCl; 5, positive control (0.33 µL DNA, 0.66 µL 1:1 NaOCl [0.1%]/thiosulfate [0.5%]); 6, 60-second exposure 0.1% NaOCl; 7, positive control (0.33 µL DNA, 0.66 µL 1:1 NaOCl [0.01%]/thiosulfate [0.05%]); 8, 60-second exposure 0.01% NaOCl. Arrow (I and II) shows reaction product of 138 bp.
In this study, a method of inactivation was developed (65°C, 2 hours) to ensure cell death without immediate cell lysis or severe DNA damage. Here, cell death was established by routine culture, confirming previous reports of lost viability of E. faecalis at temperatures above 60°C (27), with structural preservation of cells (28). DNA decay was documented by MPN-PCR analysis. While the MPN-PCR analysis provides a semiquantitative estimate of the number of 16S rRNA genetic units per sample, it does not indicate actual population sizes because of gene copy number, loss of target DNA during extraction, and PCR detection limits (29).
Although PCR detection limits of 10 cells have been reported (30), this requires ethanol precipitation, adding all extracted DNA to the PCR mixture. In the present study only a small fraction (10 µL) of the DNA eluate (200 µL) was used, partially accounting for the higher detection limit. With the same primer set, a detection limit of 6.7 × 106 ± 4.95 × 106 cells has been reported (31). Such variation reflects differences in DNA extraction efficiency and PCR reaction mixture and cycle parameters. The detection level of DNA here (approximately 1 pg) parallels other reports with 35 PCR cycles (32).
In this study, treatment with 1% NaOCl for 60 seconds destroyed amplifiable DNA. Dead cell DNA was more susceptible to NaOCl degradation presumably as a result of its more advanced state of decay. Although this finding is consistent with other reports (11, 33), previous studies did not control for inhibition of PCR by thiosulfate-inactivated NaOCl or were unable to distinguish NaOCl-based DNA destruction from inhibition of PCR by thiosulfate-inactivated NaOCl (34). Inactivation of NaOCl by sodium thiosulfate produces sodium salts such as NaCl, which inhibit DNA polymerase at elevated concentrations (35), and our results demonstrate concentration-dependent salt-induced inhibition of the PCR reaction.
Our results show that DNA can persist and be detected by PCR for at least 1 year after cell death. NaOCl efficiently destroys DNA, and our findings highlight a previously overlooked problem of inhibition of the PCR reaction by thiosulfate-inactivated NaOCl. This type of information is essential in establishing the authenticity of PCR-based isolates as active participants in the endodontic flora.
Acknowledgments
This study was supported by grants from the Australian Society of Endodontology Inc, the Australian Dental Research Foundation Inc, and University of Melbourne. Marnie Collins, Statistical Consulting Centre, University of Melbourne provided statistical support.
References
1. Sundqvist G, Figdor D. Life as an endodontic pathogen: ecological differences between the untreated and root-filled root canals. Endodontic Topics 2003;6:3–28.
2. Sundqvist G. Taxonomy, ecology, and pathogenicity of the root canal flora. Oral Surg Oral Med Oral Pathol 1994;78:522–30.
3. Sundqvist G. Bacteriological studies of necrotic dental pulps (dissertation). Umeå, Sweden: Umeå University, 1976.
4. Siqueira JF Jr, Rôças IN, Favieri A, Santos KR. Detection of Treponema denticola in endodontic infections by 16S rRNA gene-directed polymerase chain reaction. Oral Microbiol Immunol 2000;15:335–7.
5. Gonçalves RB, Mouton C. Molecular detection of Bacteroides forsythus in infected root canals. J Endod 1999;25:336–40.
6. Munson MA, Pitt-Ford T, Chong B, Weightman A, Wade WG. Molecular and cultural analysis of the microflora associated with endodontic infections. J Dent Res 2002;81:761–6.
7. O’Rourke DH, Hayes M, Carlyle S. Ancient DNA studies in physical anthropology. Annu Rev Anthropol 2000;29:217–42.
8. Bar W, Kratzer A, Machler M, Schmid W. Postmortem stability of DNA. Forensic Sci Int 1988;39:59–70.
9. Imaizumi K, Miyasaka S, Yoshino M. Quantitative analysis of amplifiable DNA in tissues exposed to various environments using competitive PCR assays. Sci Justice 2004;44:199–208.
10. Pfeiffer H, Huhne J, Seitz B, Brinkmann B. Influence of soil storage and exposure period on DNA recovery from teeth. Int J Legal Med 1999;112:142–4.
11. Ng YL, Spratt D, Sriskantharajah S, Gulabivala K. Evaluation of protocols for field decontamination before bacterial sampling of root canals for contemporary microbiology techniques. J Endod 2003;29:317–20.
12. Willson VA. Determination of available chlorine in hypochlorite solutions by direct titration with sodium thiosulfate. Industrial and Engineering Chemistry Analytical Edition 1935;7:44–5.
13. Jacob AE, Hobbs SJ. Conjugal transfer of plasmid-borne multiple antibiotic resistance in Streptococcus faecalis var. zymogenes. J Bacteriol 1974;117:360–72.
14. Sedgley CM, Nagel AC, Shelburne CE, Clewell DB, Appelbe O, Molander A. Quantitative real-time PCR detection of oral Enterococcus faecalis in humans. Arch Oral Biol 2005;50:575–83.
15. Klee AJ. A computer program for the determination of most probable number and its confidence limits. J Microbiol Methods 1993;18:91–8.
16. Klee AJ. Most probable number calculator, version 4.04. Cincinnati, OH: Risk Reduction Engineering Laboratory, United States Environmental Protection Agency, 1996.
17. Josephson KL, Gerba CP, Pepper IL. Polymerase chain reaction detection of nonviable bacterial pathogens. Appl Environ Microbiol 1993;59:3513–5.
18. Masters CI, Shallcross JA, Mackey BM. Effect of stress treatments on the detection of Listeria monocytogenes and enterotoxigenic Escherichia coli by the polymerase chain reaction. J Appl Bacteriol 1994;77:73–9.
19. Birch L, Dawson CE, Cornett JH, Keer JT. A comparison of nucleic acid amplification techniques for the assessment of bacterial viability. Lett Appl Microbiol 2001;33:296–301.
20. Sheridan GE, Szabo EA, Mackey BM. Effect of post-treatment holding conditions on detection of tufA mRNA in ethanol-treated Escherichia coli: implications for RT-PCR-based indirect viability tests. Lett Appl Microbiol 1999;29:375–9.
21. Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993;362:709–15.
22. Herrmann B, Hummel S. Introduction. In: Herrmann B, Hummel S, eds. Ancient DNA. New York: Springer-Verlag, 1994;1–12.
23. Tuross N. The biochemistry of ancient DNA in bone. Experientia 1994;50:530–5.
24. Aardema BW, Lorenz MG, Krumbein WE. Protection of sediment-adsorbed transforming DNA against enzymatic inactivation. Appl Environ Microbiol 1983;46: 417–20.
25. Lorenz MG, Wackernagel W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl Environ Microbiol 1987;53:2948 –52.
26. Deere D, Porter J, Pickup RW, Edwards C. Survival of cells and DNA of Aeromonas salmonicida released into aquatic microcosms. J Appl Bacteriol 1996;81:309–18.
27. Flahaut S, Benachour A, Giard JC, Boutibonnes P, Auffray Y. Defense against lethal treatments and de novo protein synthesis induced by NaCl in Enterococcus faecalis ATCC 19433. Arch Microbiol 1996;165:317–24.
28. van Mullem PJ, Wijnbergen M. Effect of disinfection on number and stainability of gram-positive bacteria. Int Endod J 1989;22:278–82.
29. Lozupone CA, Klein DA. Molecular and cultural assessment of chytrid and Spizellomyces populations in grassland soils. Mycologia 2002;94:411–20.
30. Molander A, Lundquist P, Papapanou PN, Dahlén G, Reit C. A protocol for polymerase chain reaction detection of Enterococcus faecalis and Enterococcus faecium from the root canal. Int Endod J 2002;35:1–6.
31. Nandakumar R, Mirchandani R, Fouad A. Primer sensitivity: can it influence the results in Enterococcus faecalis prevalence studies? Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103:429–32.
32. Palmer HM, Higgins SP, Herring AJ, Kingston MA. Use of PCR in the diagnosis of early syphilis in the United Kingdom. Sex Transm Infect 2003;79:479–83.
33. Prince AM, Andrus L. PCR: how to kill unwanted DNA. Biotechniques 1992;12: 358–60.
34. Fouad AF, Barry J. The effect of antibiotics and endodontic antimicrobials on the polymerase chain reaction. J Endod 2005;31:510–3.
35. Abu Al-Soud W, Radstrom P. Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl Environ Microbiol 1998;64:3748–53.
From the *School of Dental Science, University of Melbourne, Melbourne, Australia; †Department of Microbiology, Monash University, Melbourne, Australia; and ‡Department of Odontology/Endodontics, Faculty of Medicine, Umeå University, Umeå, Sweden.
Address requests for reprints to Dr David Figdor, 517 St Kilda Road, Melbourne, VIC 3004, Australia. E-mail address: [email protected].
0099-2399/$0 - see front matter
Copyright © 2007 by the American Association of Endodontists.
doi:10.1016/j.joen.2007.09.002